【论著】环指蛋白213基因变异相关婴儿期早发型烟雾病三例临床及遗传学特征分析
时间:2025-09-14 12:13:33 热度:37.1℃ 作者:网络
摘要:目的探讨环指蛋白213(RNF213)基因变异相关婴儿期早发型烟雾病的临床及遗传学特征。方法回顾性连续纳入2020年6月至2024年6月于华中科技大学同济医学院附属武汉儿童医院小儿神经内科收治的婴儿期早发型烟雾病患儿。收集患儿的一般及临床资料,包括性别、年龄、主诉、入院时间、现病史、个人史、家族史、神经系统体格检查、辅助检查、诊疗经过及随访情况。分析患儿及其父母基因检测结果,经生物信息学分析筛选和注释突变位点,通过Sanger测序验证可疑变异位点来源。使用分子进化遗传学分析软件MEGA(v11.0)对检出的基因错义突变位点进行氨基酸序列比对及保守性分析。基于蛋白质结构数据库获取冷冻电镜解析的蛋白三维结构,将已识别错义突变定位至该蛋白三维结构,并利用PyMOL(v2.0.6)软件进行蛋白三维结构可视化及变异位点氢键分析。结果(1)共纳入3例婴儿期早发型烟雾病患儿,男1例,女2例。3例患儿表现为癫痫发作、短暂性脑缺血发作或急性脑梗死。基因检测结果显示,3例患儿均存在RNF213基因错义突变,患儿1携带p.Arg4810Lys和p.Thr1727Met突变,均来自母亲;患儿2携带来自父亲的p.Arg4810Lys突变;患儿3携带新发突变p.Lys4136Gln,父母均为野生型。3例患儿均接受间接脑血运重建术,术后癫痫发作和脑缺血症状均逐渐缓解,末次随访改良Rankin量表评分分别为2、1、2分。(2)纳入的3例患儿RNF213蛋白的氨基酸位点苏氨酸1727(THR1727)、赖氨酸4136(LYS4136)、精氨酸4810 (ARG4810)的氨基酸位点均绝对保守。相较于野生型ARG4810,p.Arg4810Lys发生在1个α螺旋之内,氨基酸改变后内部的氢键连接减少1个;相较于野生型THR1727,p.Thr1727Met减少了1个氢键连接;相较于野生型LYS4136, p.Lys4136Gln减少了1个氢键连接,且2个α螺旋之间连接的残基均发生了改变。结论婴儿期早发型烟雾病起病隐匿,病情严重,临床表现复杂多样,易被误诊。RNF213基因p.Arg4810Lys、p.Thr1727Met和p.Lys4136Gln突变可能通过影响其蛋白质功能的分子机制参与烟雾病发病,早期RNF213基因遗传学检测有助于临床诊断。间接脑血运重建术或可使该类患儿获益。本研究结果尚需大型、前瞻性、多中心研究进一步证实。
烟雾病为罕见的慢性进展性脑血管疾病,其特征为非动脉粥样硬化性血管闭塞,主要累及双侧颈内动脉末端及其主要分支血管,并伴有颅底异常血管网形成[1]。目前烟雾病的病因尚未完全明确,遗传和环境因素均可增加其发病风险[2]。流行病学调查结果显示,儿童烟雾病高发年龄为5~9岁,成人为35~39岁[3]。潜在的遗传因素可能导致烟雾病患者起病早、病情重,表现为更广泛的血管受累范围和更严重的神经功能损伤[4]。
目前,关于婴儿期起病的烟雾病报道相对较少。笔者拟回顾婴儿期早发型(起病年龄<1岁)烟雾病患儿的病历资料并分析其基因检测结果,总结其临床及遗传学特征,以期提高临床医师对该类疾病的认识。
1 对象与方法
1.1 对象
回顾性连续纳入2020年6月至2024年6月于华中科技大学同济医学院附属武汉儿童医院小儿神经内科收治的婴儿期早发型烟雾病患儿。
纳入标准:(1)入组年龄<3岁,且为早发型(起病年龄<1岁);(2)符合《烟雾病和烟雾综合征诊断与治疗中国专家共识(2024版)》[1]关于烟雾病的诊断标准,并经头部MR血管成像(MRA)或头部DSA证实;(3)病历资料完整且行遗传学检查。
排除标准:(1)烟雾综合征(明确病因导致的类似烟雾病的脑血管改变)[1];(2)治疗依从性差;(3)失访。
本研究方案经华中科技大学同济医学院附属武汉儿童医院医学伦理委员会审核批准(伦理审批号:2024R059-E01)。所有患儿监护人签署了诊疗知情同意书。
1.2 资料收集
收集患儿的一般及临床资料,包括性别、年龄、主诉、入院时间、现病史、个人史、家族史、神经系统体格检查、辅助检查、诊疗经过及随访情况。
1.3 基因检测方法及致病性分析
采集患儿及其父母的静脉血样2ml,应用全外显子测序技术进行基因检测,经生物信息学分析筛选和注释突变位点,通过Sanger测序验证可疑变异位点来源。根据美国医学遗传学与基因组学学会(American College of Medical Genetics,ACMG)遗传变异分类标准与指南[5]对所有变异位点进行致病性评级。
1.4 生物信息学分析
使用分子进化遗传学分析软件MEGA(v11.0)对检出的基因错义突变位点进行氨基酸序列比对及保守性分析。从UniProt蛋白质数据库(https:// www.uniprot.org/)中获取蛋白的直系同源序列,涵盖人、非人灵长类、啮齿类、食肉类、偶蹄类等代表性物种。通过熵公式S= -∑nt=1P(xi)log2P(xi)比对结果的保守性,S为氨基酸位点的熵值即保守性得分,n代表该位点可能出现的氨基酸种类数,P(xi)表示第i种氨基酸在该位点出现的频率。当某位点仅有1种氨基酸存在,其频率P(xi)=1,其他氨基酸频率为0时熵值为0,表明该位点绝对保守;氨基酸种类越多,熵值越高,表明该位点保守性越低[6]。
基于蛋白质结构数据库(protein data bank,PDB;http:// www. rcsb. org/)获取冷冻电镜解析的蛋白三维结构,将已识别的错义突变定位至该蛋白三维结构,并利用PyMOL(v2.0.6)软件进行蛋白三维结构可视化及变异位点氢键分析。
2 结果
共纳入婴儿期早发型烟雾病患儿3例。
2.1 一般及临床资料
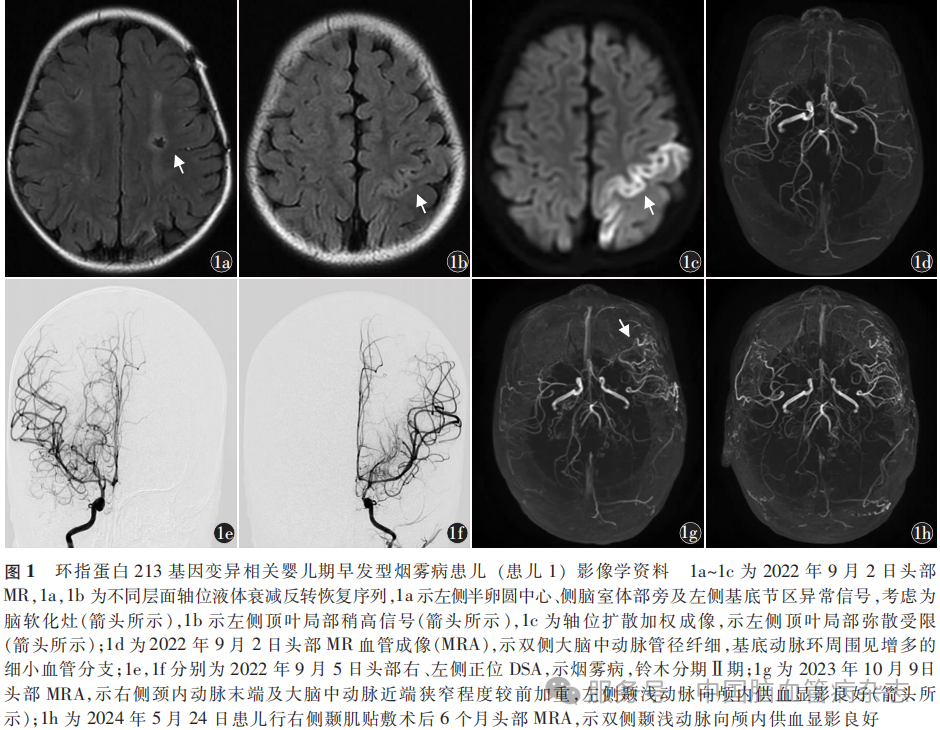
患儿1 女,1岁6个月,因“间断抽搐9个月,右侧肢体无力1d”于2022年9月2日入住华中科技大学同济医学院附属武汉儿童医院小儿神经内科。患儿自9月龄起出现发作性愣神,伴或不伴右侧肢体抖动,入院前近1个月发作频率增加,发作后偶有短暂性步态不稳。入院前1d出现持续性右侧肢体无力。个人史:患儿为第1胎第1产,足月顺产,出生体质量3500g。家族史:患儿祖父50岁时诊断为脑梗死。入院神经系统体格检查:意识清楚,头颅无畸形,颅神经检查无异常,右侧肢体肌力Ⅳ级,左侧肢体肌力Ⅴ级,右侧肌张力减低,左侧肌张力正常,双侧腱反射正常引出,双侧Babinski征阴性。营养良好,皮肤未见异常,呼吸音正常,心律齐,心脏杂音未闻及。辅助检查:2022年9月2日头部MR液体衰减反转恢复序列示左侧半卵圆中心、侧脑室体部旁及左侧基底节区异常信号,考虑为软化灶(图1a),左侧顶叶局部稍高信号(图1b),扩散加权成像示左侧顶叶局部弥散受限(图1c),考虑左侧顶叶脑梗死;头部MRA示双侧大脑中动脉管径纤细,基底动脉环周围见增多的细小血管分支(图1d),考虑烟雾病。2022年9月5日头部DSA示烟雾病,铃木分期[7]为Ⅱ期(图1e,1f)。超声心动图、心电图、胸部X线及双肾、肾血管彩色超声检查均未见明显异常。入院实验室检查同型半胱氨酸、抗中性粒细胞胞质抗体、抗心磷脂抗体免疫球蛋白G和免疫球蛋白M等均未见异常。2022年9月5日行基因检测,结果显示,患儿存在环指蛋白213(ring finger protein 213,RNF213)基因的c.14429G>A(p.Arg4810Lys)和c.5180C>T(p.Thr1727Met)错义突变,家系遗传学分析显示均来自母系遗传,两类突变在参考人群基因频率数据库(genome aggregation database, gnomAD; https:// gnomad. broadinstitute. org/)中最小等位基因频率分别为0.0006和0.0004,ACMG遗传变异分类标准与指南评级分别为可能致病及意义未明变异,患儿母亲暂无烟雾病表型。患儿存在父系来源的内皮型一氧化氮合酶基因c.2446C>T (p.Arg816Cys)杂合变异,家系共分离分析显示,患儿父亲与祖父均携带同一位点突变,构建了连续三代的父系传递链,gnomAD验证人群最小等位基因频率为0,ACMG遗传变异分类标准与指南评级为意义未明变异,关联疾病为缺血性卒中,患儿父亲无疾病表现,祖父存在早发性(50岁)缺血性卒中。住院期间给予患儿口服阿司匹林25mg/次,2次/d抗血小板聚集,左乙拉西坦口服溶液200mg/次,2次/d抗癫痫发作。患儿于2022年9月9日出院,出院时无癫痫发作,可独自行走,但跑动时步态不稳。出院后继续口服阿司匹林和左乙拉西坦口服溶液,剂量同前,规律进行康复治疗。2022年10月5日患儿因“右侧肢体乏力1d”再次入院,2022年10月14日于华中科技大学同济医学院附属武汉儿童医院神经外科行左侧颞肌贴敷术。此后患儿左侧肢体无力发作数次。2023年10月6日患儿左侧肢体无力发作持续约30min,发作停止后当日再次入院。2023年10月9日头部MRI+MRA示无新发脑梗死,右侧颈内动脉末端及大脑中动脉近端狭窄程度较前加重,左侧颞浅动脉向颅内供血显影良好(图1g)。2023年10月11日于华中科技大学同济医学院附属武汉儿童医院神经外科行右侧颞肌贴敷术。2024年5月24日复查头部MRA,示双侧颞浅动脉向颅内供血显影良好(图1h),但患儿仍存在短暂性脑缺血发作,表现为间歇性右侧肢体无力,术后1年内累计发作数次。末次随访至2025年6月,患儿规律口服阿司匹林25mg/次,2次/d抗血小板聚集,左乙拉西坦口服溶液200mg/次,2次/d抗癫痫发作,近5个月未再出现发作性肢体无力,可自主进食、行走,能执行简单指令,但存在言语不清,改良Rankin量表(mRS)评分2分。
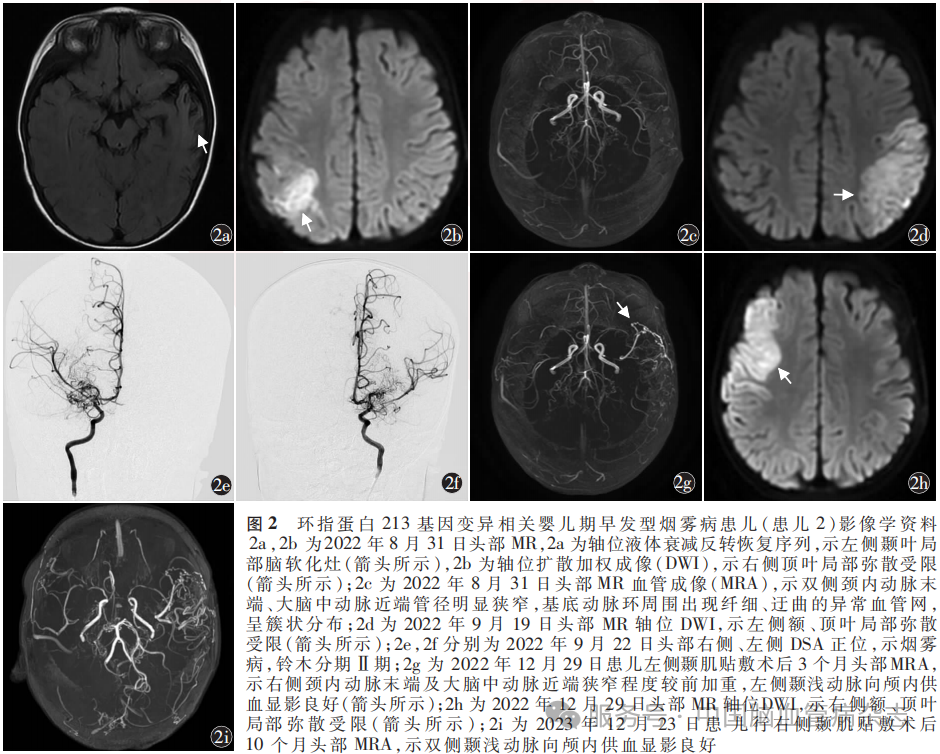
患儿2 男,1岁6天,因“发作性抽搐7周余,加重伴左侧肢体乏力9h”于2022年8月29日入住华中科技大学同济医学院附属武汉儿童医院小儿神经内科。患儿入院前7周余无诱因出现抽搐,表现为口唇发绀伴咂嘴、吞咽动作,四肢僵直,持续10余秒后自行缓解,每日发作数次,3d后发作停止。入院前9h患儿连续抽搐2次,均表现为意识丧失,左上肢节律性抖动,持续1min后停止,发作后出现持续性左侧肢体乏力。个人史:患儿为第3胎第2产,足月顺产,出生体质量3500g,其母亲第2胎于孕7周时胚胎停育流产。患儿语言及运动功能发育未见明显异常。家族史:患儿祖父(发病年龄52岁)和外祖父(发病年龄53岁)患卒中,具体病因未明确。入院神经系统体格检查:意识清楚,左侧肢体肌力Ⅳ级,左侧肢体肌张力稍高,左侧腱反射稍活跃,左侧Babinski征阳性。心、肺、腹及颅神经检查无异常。入院实验室检查:同型半胱氨酸水平15.20μmol/L(正常范围:<10.00μmol/L),其他血液学、血管炎筛查等实验室检查指标均未见明显异常。2022年8月31日头部MRI示右侧顶叶脑梗死,左侧颞叶软化灶伴脑萎缩(图2a,2b);头部MRA示双侧颈内动脉末端、大脑中动脉近端管径明显狭窄,基底动脉环周围出现纤细、迂曲的异常血管网,呈簇状分布(图2c),考虑烟雾病。超声心动图、心电图、胸部X线及双肾和肾血管彩色超声未见异常。2022年9月5日行基因检测,结果显示,患儿存在RNF213基因c.14429G>A(p.Arg4810Lys)错义突变,家系共分离分析结果显示,患儿父亲与祖父均携带该位点突变,患儿父亲无临床表现,但祖父52岁出现早发性卒中;叶酸代谢基因分析显示,亚甲基四氢叶酸还原酶基因c.677C>T和c.1298A>C基因型分别为C等位基因的纯合子和A等位基因的纯合子,5-甲基四氢叶酸-高半胱氨酸甲基转移酶还原酶基因的C.66A>G基因型为A等位基因的纯合子,均为低风险,酶活性正常。给予患儿口服叶酸片0.2mg/次,2次/d,维生素B6片5mg/次,2次/d,甲钴胺片0.25mg/次,2次/d,1个月后复查同型半胱氨酸,恢复正常水平后停用;给予口服阿司匹林片25mg/次,2次/d抗血小板聚集,左乙拉西坦口服溶液200mg/次,2次/d抗癫痫发作。2022年9月5日出院,出院时患儿左侧肢体肌力恢复至Ⅴ级,口服左乙拉西坦溶液后未再出现明显癫痫发作。2022年9月18日患儿因“右侧肢体无力1d”再次入院,右侧肢体肌力Ⅱ级,2022年9月19日头部MRI示左侧额、顶、枕叶新发梗死灶(图2d)。2022年9月22日于华中科技大学同济医学院附属武汉儿童医院神经外科行左侧颞肌贴敷术,术中DSA示烟雾病,铃木分期Ⅱ期(图2e,2f)。术后继续口服阿司匹林片25mg/次,2次/d抗血小板聚集。2022年12月27日患儿因“左侧肢体乏力1d”再次入住华中科技大学同济医学院附属武汉儿童医院神经外科,神经系统体格检查左侧肢体肌力Ⅳ级。2022年12月29日头部MRA示右侧颈内动脉末端及大脑中动脉近端狭窄程度较前加重,左侧颞浅动脉向颅内供血显影良好(图2g),头部MRI示右侧额、顶叶新发梗死灶(图2h)。患儿起病3d后肢体力量恢复,家属要求择期手术。2023年2月13日于华中科技大学同济医学院附属武汉儿童医院神经外科行右侧颞肌贴敷术。术后继续口服阿司匹林片25mg/次,2次/d抗血小板聚集,左乙拉西坦口服溶液200mg/次,2次/d抗癫痫发作。术后未再发卒中事件或癫痫发作。2023年12月23日复查头部MRA示双侧颞浅动脉向颅内供血显影良好(图2i)。第2次术后1年停用阿司匹林片和左乙拉西坦片口服溶液。末次随访至2025年7月,患儿可完成快走和跑动,但左侧下肢存在轻度跛行,语言及智能发育正常,mRS评分1分。
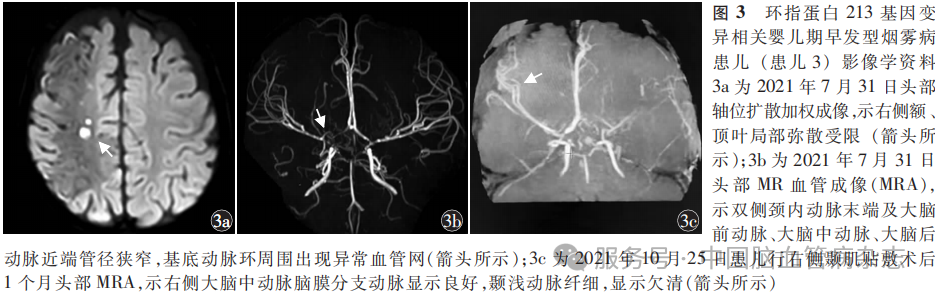
患儿3 女,9月龄,因“间断抽搐伴左侧肢体活动减少3d”于2021年7月26日由外院转入华中科技大学同济医学院附属武汉儿童医院小儿神经内科。入院前3d患儿无诱因出现抽搐,表现为意识水平下降,左侧肢体及左侧嘴角节律性抽搐,首次发作持续约30min,缓解后左侧肢体活动减少。后于当地医院住院期间仍有抽搐发作,遂转入华中科技大学同济医学院附属武汉儿童医院小儿神经内科。个人史:患儿为第1胎第1产,足月顺产,出生体质量2600g;大运动滞后于同龄儿童。三代家族史无遗传病、慢性病或早发疾病。入院神经系统体格检查:意识清楚,左侧肢体肌力Ⅲ级,左侧肢体肌张力增高,左侧膝反射活跃,左侧Babinski征阳性。皮肤无异常,心、肺、腹体格检查无异常。辅助检查:2021年7月31日头部MRI示右侧额、颞、顶叶脑梗死(图3a);头部MRA示双侧颈内动脉末端及大脑前动脉、大脑中动脉、大脑后动脉近端管径狭窄,基底动脉环周围出现异常血管网(图3b),考虑烟雾病。2021年7月31日超声心动图示左心室心尖部局部心肌结构疏松,左心室壁厚度≥5mm,考虑肥厚型心肌病。入院实验室检查:肝功能检测示丙氨酸氨基转移酶162U/L(正常范围:7 ~ 45 U/L),天冬氨酸氨基转移酶114 U/L (正常范围:10 ~ 40 U/L),1周后复查丙氨酸氨基转移酶降至62 U/L,天冬氨酸氨基转移酶降至78 U/L;血、尿代谢筛查、血液学和血管炎等相关实验室检查均未见明显异常。2021年8月1日行基因检测,结果显示,患儿存在RNF213基因c.12406A>C(p.Lys4136Gln)杂合突变,家系遗传学分析显示患儿父母在该位点均为野生型。予以口服阿司匹林片25mg/次,2次/d抗血小板聚集和左乙拉西坦口服溶液150mg/次,2次/d抗癫痫发作治疗。服用左乙拉西坦溶液2d后未再抽搐,肢体自主活动增多,于2021年8月2日出院。2021年9月14日于外院行右侧颞肌贴敷术,术后患儿每年癫痫发作≤2次,继续口服阿司匹林片(25mg/次,2次/d)抗血小板聚集,左乙拉西坦口服溶液(150mg/次,2次/d)抗癫痫发作。2021年10月25日复查头部MRA,示右侧大脑中动脉脑膜分支动脉显示良好,颞浅动脉纤细,显影欠清(图3c),双侧颈内动脉C6、C7段狭窄,双侧大脑中动脉M1段狭窄,双侧大脑前动脉纤细。末次随访至2025年7月,患儿可独立行走及自主进食,但左侧下肢跛行明显,无法完成跑步及快走动作,可简单交流,可理解指令并说简单句,术后未再发卒中,近18个月无癫痫发作。mRS评分2分。术后1年停用阿司匹林片,继续口服左乙拉西坦溶液抗癫痫治疗。
2.2 基因检测结果及致病性变异分析
3例婴儿期早发型烟雾病患儿及其父母RNF213基因突变位点测序结果见图4。

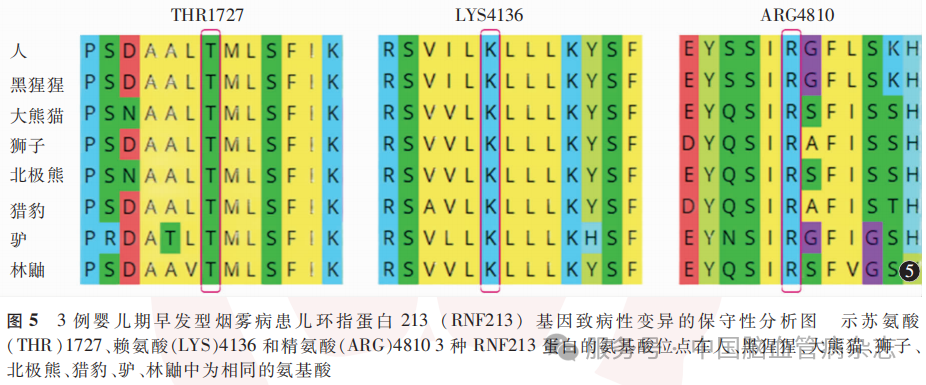
采用MEGA(v11.0)软件对检出的基因错义突变位点进行氨基酸序列比对及保守性分析,纳入的3例患儿RNF213蛋白的氨基酸位点苏氨酸1727(threonine 1727,THR1727)、赖氨酸4136(lysine 4136,LYS4136)、精氨酸4810(arginine 4810,ARG4810)在人、黑猩猩、大熊猫、狮子、北极熊、猎豹、驴、林鼬中为相同的氨基酸(图5)。3个不同氨基酸位点均仅存在1种氨基酸,熵值为0,即以上3个错义突变的氨基酸位点均绝对保守。
采用PyMol(v2.0.6)软件对人类RNF213基因蛋白质三维结构可视化,并分析纳入的3例患儿RNF213蛋白变异位点氢键。人类RNF213基因蛋白质三维模型中野生型ARG4810、THR1727和LYS4136氨基酸在蛋白质结构中的位置见图6a。相较于野生型ARG4810(图6b),p.Arg4810Lys发生在1个α螺旋之内,氨基酸改变后内部的氢键连接减少1个(图6c)。相较于野生型THR1727(图6d),p.Thr1727Met减少1个氢键连接(图6e)。相较于野生型LYS4136(图6f),p.Lys4136Gln减少1个氢键连接,且2个α螺旋之间连接的残基均发生了改变(图6g)。
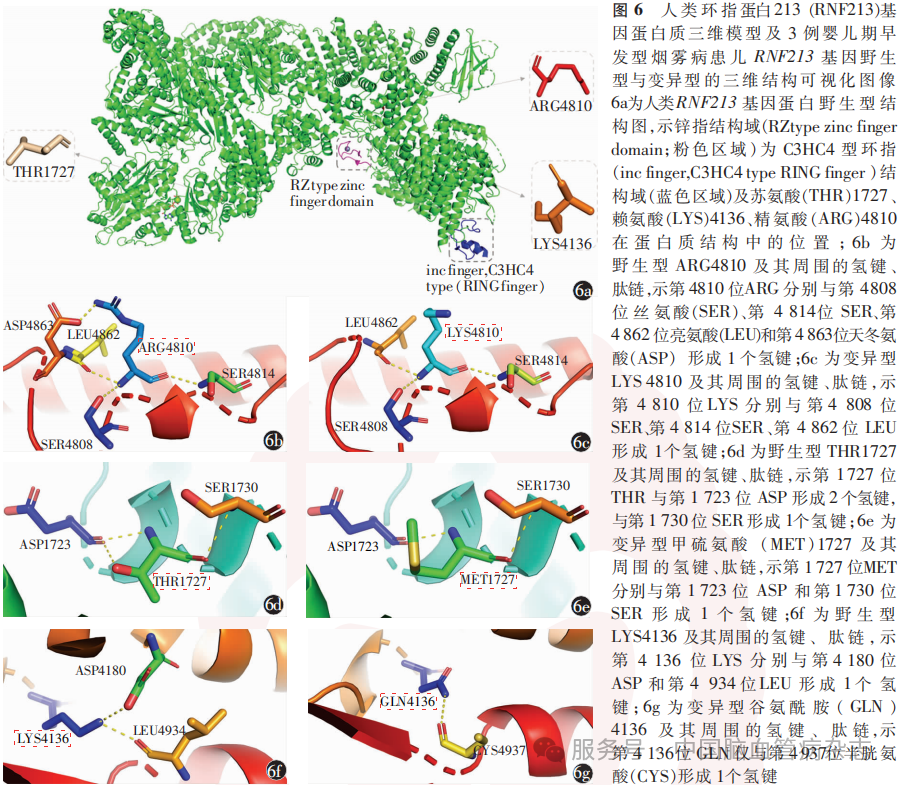
3 讨论
烟雾病于1969年由日本研究者首次发现并报道[8],其在东亚人群中发病率高,且具有家族聚集性,其致病机制与遗传因素密切相关[1]。烟雾病相关遗传学是该领域的研究热点,目前以RNF213基因的研究最为深入。RNF213基因位于染色体17q25.3,其编码的环指蛋白是具有泛素连接酶活性和腺苷三磷酸酶活性的大分子蛋白[9]。RNF213基因的突变可能影响血管生成、免疫活动以及内皮功能,这些机制是烟雾病病理学和疾病进展的基础[10]。在RNF213基因上的致病性突变多位于羧基端,包括环指(really interesting new gene, RING)、AAA+超家族[壳区;AAA + superfamily domain (shell region), Shell]、AAA+超家族[核心区;AAA + superfamily domain (core region), Core]等结构域[11]。p.Arg4810Lys位于RNF213基因的Core结构域,该突变在中国、日本、韩国烟雾病患者中的携带率分别为23%(12/52)、90%(145/161)和79%(30/38)[9]。日本人群p.Arg4810Lys的次要等位基因频率为1.36%(40/2948)[12],但日本2003年烟雾病的患病率和年发病率分别为6.03/10万和0.54/10万[13],烟雾病的患病率和年发病率均低于变异基因的携带率,表明该突变具有低外显率的特点。一项研究显示,在168例携带p.Arg4810Lys突变的烟雾病患者中,15例患者为p.Arg4810Lys纯合突变,其中9例患者于4岁前确诊;该研究对照组和未患病父母中无纯合突变,无法计算优势比,但通过计算得出,携带p.Arg4810Lys纯合突变时,烟雾病发病率的95%CI为0.78~1.00[14]。
截至2025年7月,检索人类基因突变数据库(http:// www. hgmd. cf. ac. uk/ ac/index. php)和ClinVar数据库(https:// www. ncbi. nlm. nih. gov/ clinvar/)均未见收录本研究报道的患儿3所携带的p.Lys4136Gln突变。本研究结果显示,p.Lys4136Gln突变在进化上高度保守,蛋白质三维结构模型分析显示,该突变不仅导致氢键连接数量发生变化,与之相互作用的残基也均发生改变,且这些变化发生在2个α螺旋之间,提示该突变可能对RNF213基因编码蛋白质的结构与功能产生干扰。p.Lys4136Gln突变位点位于羧基端的Shell结构域内,邻近RING和Core结构域,三者共同构成E3泛素连接酶功能模块[15]。RING和Shell结构域突变可降低E3泛素连接酶活性[16],而Shell结构域4114至4146氨基酸区域的部分新发突变则可通过影响锌离子结合结构,增加烟雾病易感性[17]。有研究显示,Shell结构域内突变的烟雾病患者发病早,临床表现相对严重[18],提示各结构域突变对烟雾病发病临床特点的影响不同。Pinard等[19]报道了3例存在RING和Shell结构域突变的烟雾病患儿,并回顾了既往研究报道的携带p.His4014Asn[20]、p.Cys4017Ser[21]、p.Lys4115del[22]及p.Ser4118Phe[23]变异的烟雾病患儿各1例,7例患儿均为3岁前起病,其中4例合并颅外动脉病变,1例出现扩张型心肌病,2例出现非慢性肝炎相关的氨基转移酶升高,经血管重建术治疗后,部分患者可获得生活自理能力。既往有研究分别报道了携带p.Asp4122Val[20]、p.Ser4118Phe[23]及Pro3996_ Cys3997delinsGlyLeuGly[19]的3例烟雾病患儿,病程中均出现一过性氨基转移酶升高,其中,携带p.Ser4118Phe变异的患儿在4月龄时接受了肝脏病理检查,结果显示存在脂肪滴和细胞型胆固醇晶体,提示病程中的一过性氨基转移酶升高可能与胆固醇酯储存疾病有关,但后续全外显子检查未见可解释氨基转移酶升高的相关致病变异[23]。本研究中,携带p.Lys4136Gln突变的患儿3也存在氨基转移酶升高,但在短期内下降。Pinard等[19]认为,位于RNF213蛋白RING和Shell这两个区域内或其附近的突变可能导致尚未明确的肝脏病变。
本研究通过PyMol软件对p.Thr1727Met和周围的氢键、肽链结构进行分析,结果显示,相对于野生型THR1727,该突变减少1个氢键连接。目前关于p.Thr1727Met突变的研究较少。一项研究显示,在255例中国烟雾病患者中,80例携带p.Arg4810Lys突变,其中29例(36.3%)同时携带p.Thr1727Met突变,与健康对照组(300名)相比,烟雾病患者中p.Arg4810Lys和p.Thr1727Met突变率均更高(均P<0.01),但p.Thr1727Met突变未显示出对烟雾病临床表现严重程度及起病年龄的影响[18]。上述研究结果提示,p.Thr1727Met突变对蛋白质三维结构的稳固性和功能影响可能较小。
目前,国内外关于婴儿期早发型烟雾病患儿的临床报道较为少见。本研究中的3例患儿均出现缺血性卒中和癫痫发作,与既往研究报道一致[24-25]。此外,婴儿期早发型烟雾病通常起病隐匿,在发病早期易被误诊为惊厥发作或行为异常,导致延误诊断及干预时机。本研究中患儿1和患儿2在就诊时影像学检查均提示存在脑软化灶,结合病史推测为既往脑缺血事件引起,其中患儿1症状出现至确诊时间长达9个月。
本研究中,患儿2存在同型半胱氨酸轻度升高,且其叶酸代谢相关基因多态性分析提示低风险。经叶酸及维生素B6补充治疗后,该患儿同型半胱氨酸水平恢复正常。高同型半胱氨酸血症与缺血性卒中、冠心病等心脑血管疾病密切相关[26],其引起的代谢紊乱可损伤内皮细胞调节血管舒张的功能,诱发炎症反应,加速内皮细胞凋亡,同时抑制一氧化氮的合成,影响血管的功能和结构[27],这些病理改变协同参与烟雾病的发生与发展。一项纳入1492例烟雾病患者的全基因组关联研究结果显示,烟雾病患者高同型半胱氨酸血症易受遗传因素影响,且其在早发型(<18岁)烟雾病中的遗传效应强于晚发型(≥18岁)[28]。另一项研究显示,相较于健康对照组(88名),出血性(78例)或缺血性(224例)烟雾病患者的血清同型半胱氨酸水平均升高(均P<0.05),该研究还构建了以同型半胱氨酸为关键决定因素的基于蛋氨酸循环相关代谢物的风险模型,结果显示,该模型与烟雾病发病独立相关(OR=7.253,95%CI:3.380~15.566, P< 0.01)[29]。
本研究中患儿1及其父亲、祖父均携带内皮型一氧化氮合酶基因的p.Arg816Cys变异,其父亲无临床表现,但祖父出现早发性卒中。内皮型一氧化氮合酶基因编码的内皮型一氧化氮合酶是血管稳态的关键调节因子,其功能缺陷导致的一氧化氮合成不足可引起血管舒缩功能障碍和血管重塑[30]。2023年Guey等[31]首次提出内皮型一氧化氮合酶双等位基因缺失可致烟雾病。有研究进一步证实,敲除RNF213基因可间接导致小鼠的一氧化氮合成下降[32],提示RNF213基因与内皮型一氧化氮合酶基因的功能可能具有协同效应。
综上所述,本研究总结分析了单中心诊断的3例婴儿期早发型烟雾病患儿,并对其突变位点的蛋白质三维结构进行了分析。受本研究样本量小的局限,此类患儿的临床及遗传学特征仍需通过大样本、多中心研究进一步证实。

