Circ Res 杨宝峰/吕延杰/潘振伟教授团队揭示E3泛素连接酶接头蛋白SPOP通过泛素化降解TEFB导致心肌肥厚和心力衰竭
时间:2025-09-25 15:39:10 热度:37.1℃ 作者:网络
心力衰竭是人类死亡的重要原因,其发病率和死亡率逐年攀升。尽管现有治疗手段不断进步,但多数患者预后仍不理想。心肌肥厚是心力衰竭的病理基础,也是心衰防治的重要干预窗口。病理性心肌肥厚是心脏在高血压、瓣膜病、冠心病等疾病条件下发生的适应性重构反应。由于心肌细胞缺失增殖能力,心脏主要通过细胞肥大、室壁增厚提高心输出量维持心脏功能。然而,长期的肥厚效应会导致心肌耗氧增加、血管密度下降,引发心肌缺血、凋亡和间质纤维化,最终导致心室顺应性降低、收缩功能下降,进展为心力衰竭甚至猝死。
蛋白质合成与降解失衡是多种心脏疾病病理过程的核心机制,泛素-蛋白酶体系统是蛋白质降解的重要通路,对维持心脏功能至关重要。斑点状锌指结构蛋白(SPOP)是Cul3-RING E3泛素连接酶的接头分子,直接识别并招募底物到E3泛素连接酶,标记泛素并通过蛋白酶体降解底物分子。SPOP底物的共同特点为含有5个残基的φ-π-S-S/T-S/T (φ-非极性;π-极性) SPOP结合基序,因其在癌症中的“重要调控作用”备受关注。
2025年9月15日,寒地心血管病全国重点实验室、哈尔滨医科大学药学院杨宝峰院士/吕延杰/潘振伟教授团队在Circulation Research杂志发表了题为“SPOP is a key trigger of pathological cardiac hypertrophy and heart failure”的研究论文,揭示了SPOP通过调控心肌细胞的线粒体自噬导致心肌肥厚和心力衰竭,而SPOP抑制剂则减轻压力负荷导致的小鼠心肌肥厚和心脏功能障碍,提高存活率,为心肌肥厚和心力衰竭的临床治疗提供潜在治疗靶点。

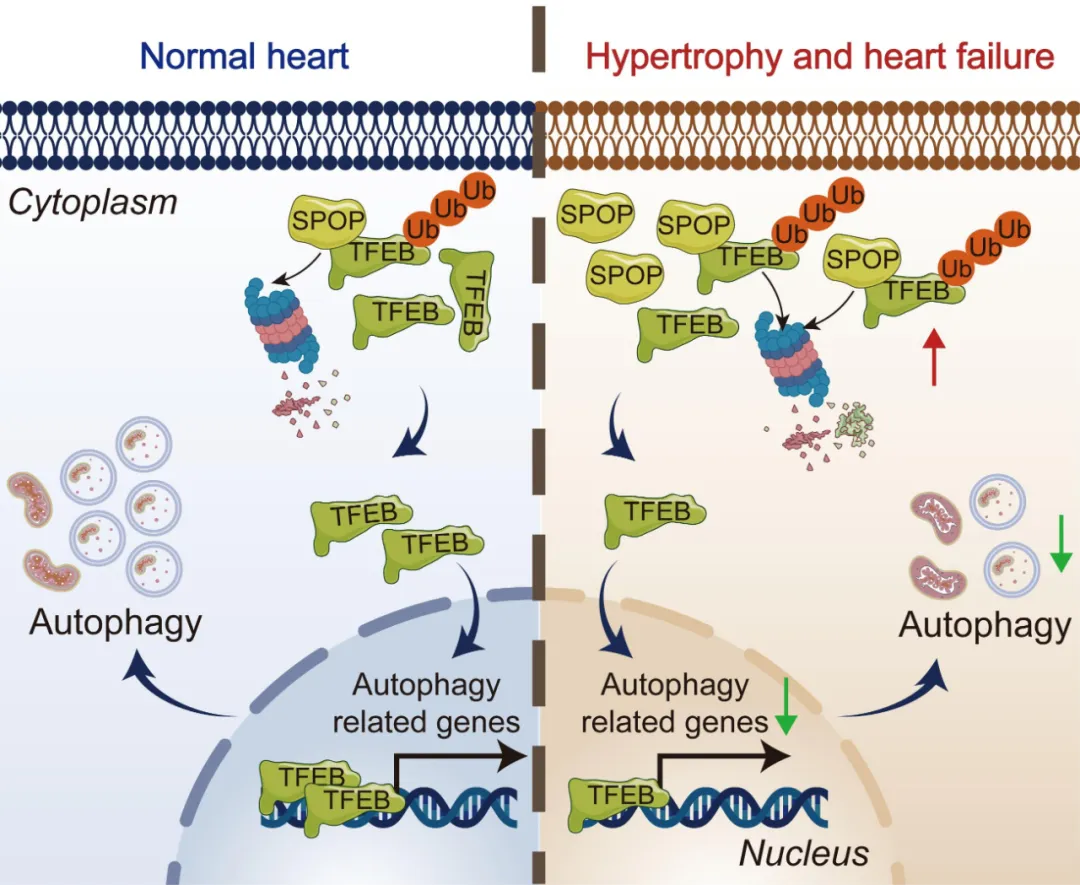
首先,团队发现SPOP在心衰患者和主动脉缩窄术(TAC)诱导的压力负荷小鼠心脏中表达显著升高,SPOP在心肌细胞中的表达丰度显著高于心脏其他类型细胞,且在压力负荷小鼠的心肌细胞中表达显著增加。心肌细胞特异性过表达SPOP (cKI)引起小鼠自发性心肌肥厚、心肌纤维化和心脏功能障碍,甚至死亡。
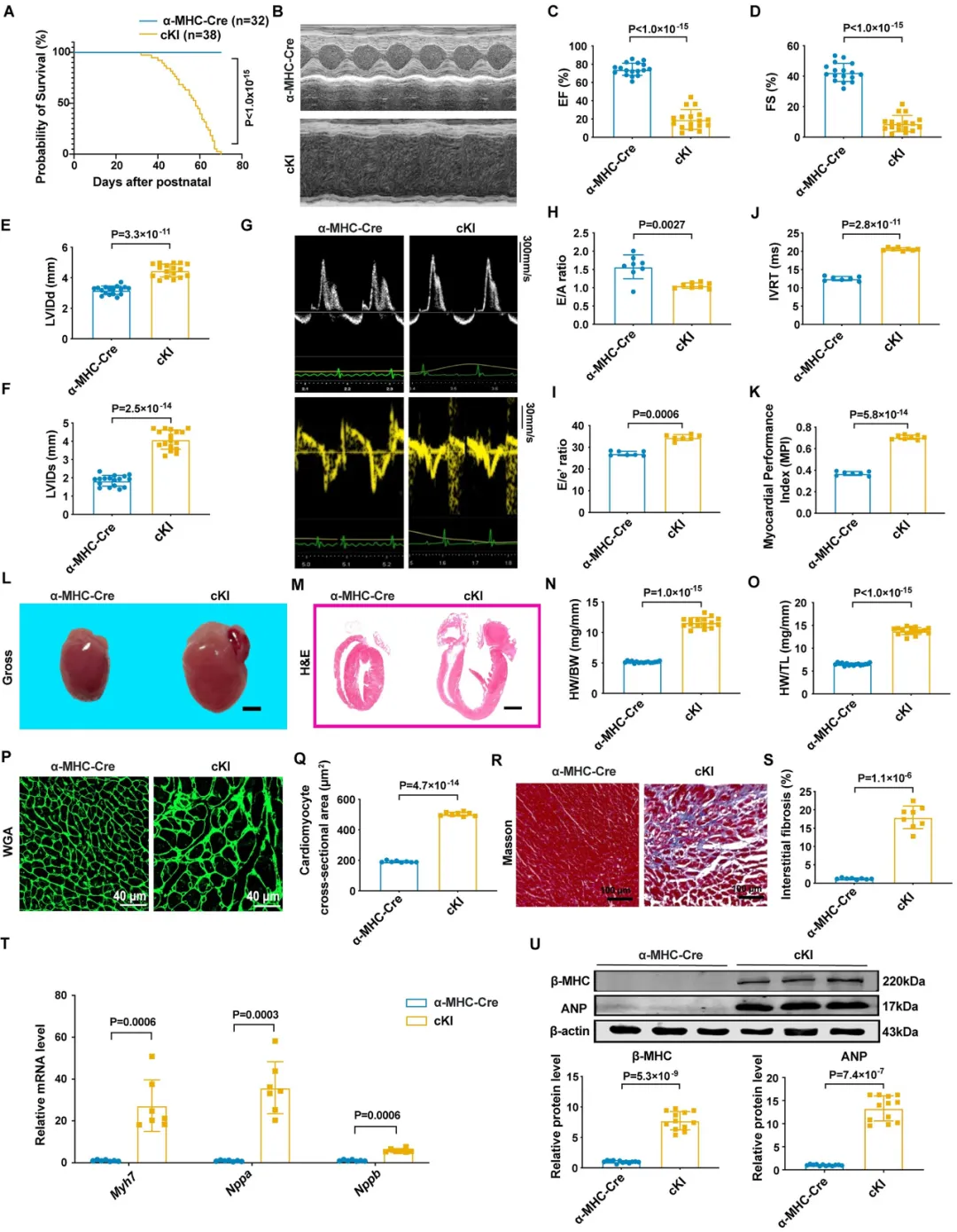
而心肌细胞特异性敲除SPOP (cKO)可减轻TAC术引起的左心室扩张和心功能障碍,减轻心肌细胞肥大及心肌纤维化,降低心肌肥厚标志物(β-MHC和ANP)的表达,显著提高小鼠存活率,证明抑制SPOP具有心脏保护作用。
在机制方面,研究者采用RNA-seq和蛋白组学联合分析发现,SPOP调控心脏自噬、溶酶体和线粒体自噬通路,过表达SPOP抑制心肌细胞自噬。为了筛选SPOP调控自噬的靶点,团队采用免疫沉淀联合质谱分析,发现转录因子EB (TFEB)是自噬和溶酶体通路里唯一含有SPOP结合基序的底物分子,SPOP与TFEB存在相互结合和共定位关系,过表达SPOP促进TFEB泛素化降解,而抑制SPOP则作用相反。
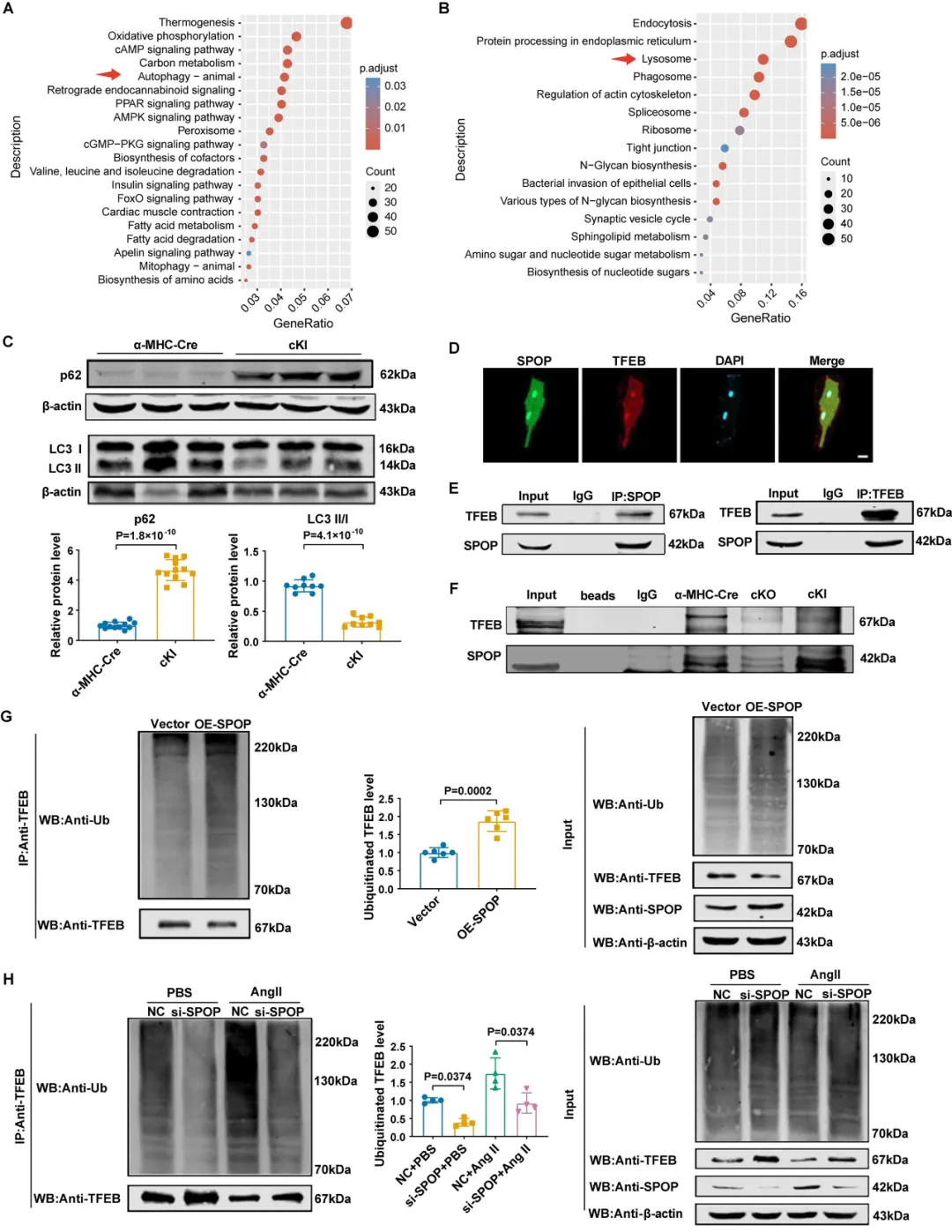
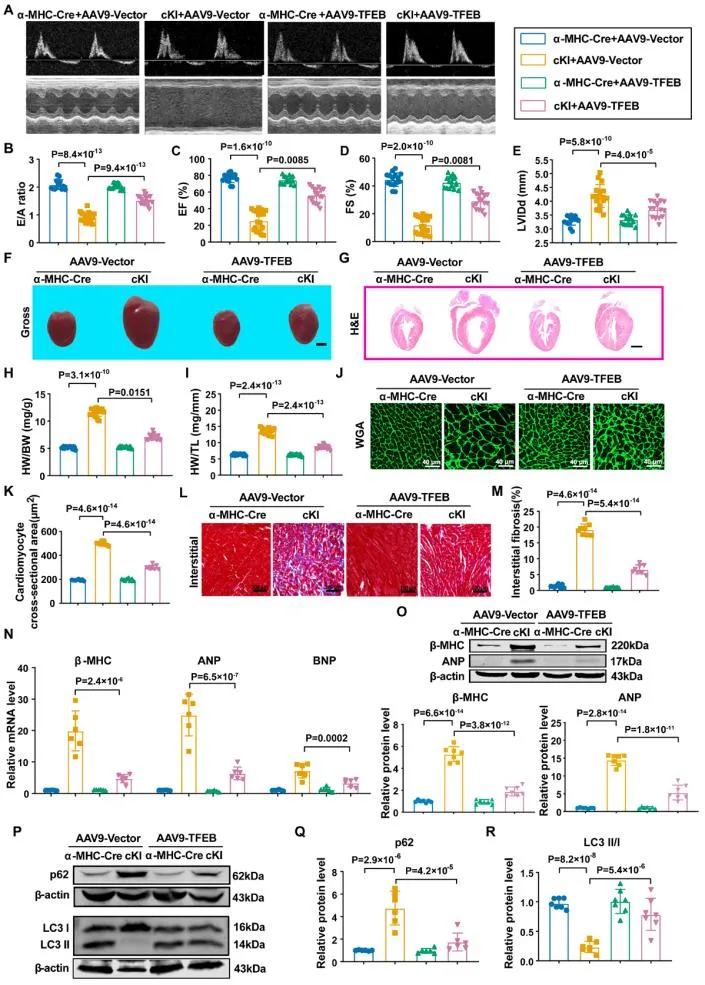
靶向心肌细胞的AAV9腺相关病毒介导的TFEB表达可显著改善SPOP过表达小鼠心脏功能、减小心脏体积、减轻纤维化和自噬水平,并提高小鼠存活率,表明TFEB是SPOP调控心脏肥大及病理性重构的关键下游分子。
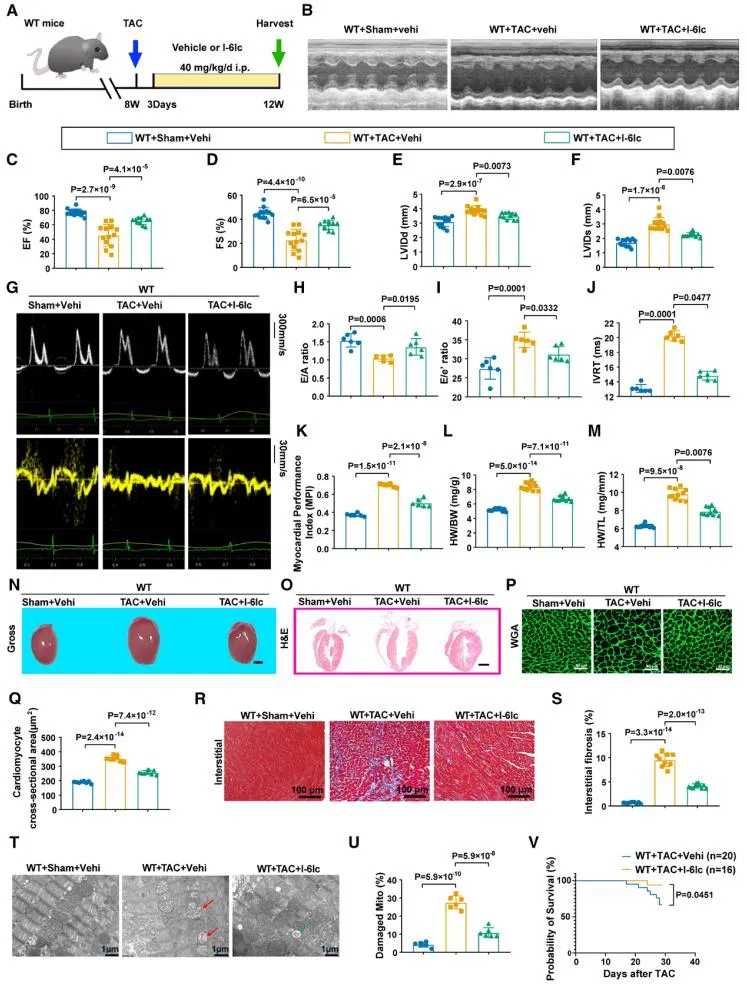
为进一步探讨SPOP的药理学干预潜力,研究者使用SPOP特异性抑制剂I-6lc处理TAC小鼠,发现SPOP抑制剂I-6lc可改善TAC小鼠的心脏功能,减轻心脏肥厚和心肌纤维化,线粒体结构和自噬功能恢复(受损线粒体减少,自噬相关基因表达上调)。并且I-6lc具有良好的安全性,具有临床应用潜力。
综上所述,本研究发现SPOP通过直接结合TFEB并促进其泛素化降解,抑制自噬效应,导致受损线粒体积累和能量代谢紊乱,促进心肌肥厚和心力衰竭。尤为重要的是,特异性SPOP抑制剂能显著减轻并改善心肌肥厚和心力衰竭。这些发现扩展了对SPOP功能的认知及其作为防治心脏肥厚和心力衰竭新靶点的潜力。
此前,团队已在Cell Chemical Biology杂志发表研究,首次揭示SPOP通过调控心脏成纤维细胞中RACK1蛋白促进心肌梗死后纤维化,本研究进一步拓展了SPOP在心脏疾病中的功能,形成“SPOP调控心血管疾病”的系列成果,扩展了SPOP对心血管疾病的调控作用和分子机制。
寒地心血管病全国重点实验室、哈尔滨医科大学药学院杨宝峰院士、吕延杰教授和潘振伟教授为论文的共同通讯作者。吴昊博士和庄宇婷副研究员为本研究的共同第一作者,哈尔滨医科大学张洋教授、杨莹副教授和李嘉亮助理研究员为本研究提供支持。该研究获得国家自然科学基金(重点项目、联合基金和面上项目)及中国科学技术部国家重点研发计划等项目支持。
原文链接:
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.125.326129

