脱水性遗传性口型红细胞增多症最初“误诊”为骨髓增生异常综合征,WES检出PIEZO新发复合杂合变异明确诊断
时间:2025-09-26 12:10:35 热度:37.1℃ 作者:网络
脱水性遗传性口形红细胞增多症(DHS)是一种罕见的常染色体显性遗传性先天性非免疫性溶血性贫血,由PIEZO1基因的致病性变异引起。其临床表现常与其他血液系统疾病重叠,导致诊断困难和潜在的管理不当。一名 22 岁男性患者因7年贫血病史就诊,最初因骨髓增生活跃和骨髓增生异常综合征(MDS)样特征被误诊为MDS。随着时间推移,其病情进展并出现严重并发症——脑静脉窦血栓形成(CVST)。全外显子测序(WES)检测发现了新发复合杂合PIEZO1变异:NM_001142864.4:c.6622A>G(p.Ile2208Val)和NM_001142864.4:c.3160C>A(p.Leu1054Met)。这些发现证实了DHS的诊断。患者接受了异基因造血干细胞移植(allo-HSCT),血液学异常和症状得到缓解。本病例强调了在不明原因贫血和MDS样特征患者中,尤其是伴有血栓并发症时,考虑DHS的重要性。它还突出了基因检测在诊断罕见遗传性贫血中的关键作用,并表明allo-HSCT在特定病例中可作为治愈性治疗。
背 景
脱水性遗传性口形红细胞增多症(DHS;OMIM:#194380)是一种常染色体显性遗传性先天性非免疫性溶血性贫血,其特征是存在口形红细胞——即呈现独特口状形态的异常红细胞(RBC)。由于DHS罕见,其患病率尚不明确,据估计,具体因研究人群而异,在人群中的发病率约为1/8000至1/50000。DHS主要由PIEZO1基因的变异引起,该基因编码一种机械敏感性离子通道,对维持红细胞内的细胞离子平衡至关重要。PIEZO1的致病性变异常导致功能获得性通道病,引起通道失活延迟及对单价阳离子(尤其是钠和钙)的通透性增加。这种调节异常最终会破坏红细胞正常的渗透调节机制,导致口形红细胞的形成。
DHS常因临床特征和实验室检查结果与其他溶血性疾病重叠而被误诊为其他溶血性疾病。DHS与多种溶血性贫血均表现为非特异性症状,如贫血、黄疸和脾肿大,仅根据临床表现难以区分。此外,DHS特征性的口形红细胞可能被误认为其他疾病(如遗传性球形红细胞增多症或骨髓增生异常综合征[MDS])中出现的异常红细胞形态。红细胞指数的细微差异及其他细胞减少症的存在进一步使诊断过程复杂化。因此,临床医生可能会忽视DHS的特定遗传基础,转而依赖更常见的诊断。这种误诊可能导致不恰当的治疗和延误正确诊断,进而加重症状并增加医疗成本。值得注意的是,脾切除术常用于免疫介导性溶血性贫血的症状管理,但在DHS中无效,且与血栓栓塞并发症风险显著增加相关。因此,对疑似病例进行全面评估和基因检测对于确保DHS的准确诊断至关重要。
本文报告一名 22 岁男性患者,该患者因骨髓增生活跃(BM)表现和重叠的临床症状掩盖了DHS的准确诊断,被误诊为MDS长达 7 年。这种误诊导致其病情逐渐恶化,症状从最初的贫血发展为脑静脉窦血栓形成(CVST)——一种可能危及生命的并发症。通过全外显子测序(WES),研究人员在该患者中发现了新发复合杂合PIEZO1变异,包括母源遗传的NM_001142864.4:c.6622A>G(p.Ile2208Val)和父源遗传的NM_001142864.4:c.3160C>A(p.Leu1054Met)。患者最终被确诊为DHS,并通过骨髓移植治愈。
病 例
既往病史中有MDS样特征
2023 年 3 月,一名有 7 年贫血病史的 22 岁男性因健康状况前往宁波大学附属第一医院血液科寻求二次诊疗意见。患者 15 岁时,在常规体检中发现患有贫血(血红蛋白 89 g/L,平均红细胞体积[MCV] 108 fL)。尽管进行了进一步检查,但未发现这些异常的明显潜在病因。患者最初被给予叶酸和维生素B12补充治疗,但其症状改善甚微。
在随后的 3 个月内,患者病情逐渐加重,疲劳感加剧并反复出现黄疸。他随后在当地医院接受了全面的血液学检查,初步诊断为骨髓增生异常综合征(MDS)。外周血涂片显示小部分(9%)红细胞呈椭圆形。血清铁蛋白、叶酸和维生素B12水平均在正常范围内。其他检查包括抗人球蛋白试验(库姆斯试验)、血红蛋白电泳、高铁血红蛋白还原试验(MRT)和渗透脆性试验(OFT),结果均正常。骨髓穿刺显示骨髓增生活跃,粒红比(M/E)升高至 5.54:1。髓系细胞增殖活跃,尤其是中性粒细胞前体细胞比例增加。骨髓中细胞形态总体正常,未见异型细胞特征。骨髓穿刺标本的多参数流式细胞术检测显示原始细胞 <1%,免疫表型显示CD34、CD38、CD58、髓过氧化物酶(MPO)和HLA-DR阳性。骨髓细胞染色体分析显示正常男性核型(46, XY),针对MDS常见染色体异常的荧光原位杂交(FISH)结果为阴性。基于这些发现,当地医院怀疑为MDS,但未启动进一步治疗。
CVST进展
此次就诊前 10 个月,患者开始出现不明原因的左侧颞部持续性疼痛,伴有间歇性头晕和出汗。随后,患者再次就诊于此前的同一家医院。全血细胞计数(CBC)显示严重贫血(红细胞计数 1.62×10¹²/L,血红蛋白 60 g/L),血小板计数轻度升高,为 366×10⁹/L。凝血参数未检测。影像学检查(包括磁共振成像[MRI]和脑血管造影)显示脑静脉窦血栓形成(CVST)证据,双侧横窦及双侧乙状窦可见多发血栓性病变。确诊CVST后,患者接受了抗凝治疗、导管导向血管内溶栓治疗、球囊静脉成形术及对症治疗。经过 10 天的强化治疗后,其头痛和头晕症状缓解,神经功能未受影响。出院时,患者被开具了长期口服药物,包括华法林、琥珀酸亚铁、叶酸和维生素C。
DHS的最终诊断
为查明其持续症状及复杂临床表现的确切原因,患者前往宁波大学附属第一医院寻求进一步诊治。入院时体格检查显示苍白(结膜及手掌)、轻度巩膜黄染,以及心动过速(心率 112 次/分钟)。腹部触诊及超声检查未发现肝脾肿大。神经系统检查未见局灶性缺损或脑膜刺激征。全面回顾患者病史后,怀疑存在可解释其病情的潜在血液系统异常,遂决定对其进行详细的重新评估。
与既往结果一致,血液实验室检查显示严重贫血(表1)。凝血功能检查显示凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、凝血酶时间(TT)及国际标准化比值(INR)升高,其他凝血指标均在正常范围内(表1)。值得注意的是,评估时患者正在接受抗凝治疗,这可能对凝血参数产生了影响。值得注意的是,外周血涂片再次发现红细胞形态异常(图1A)。骨髓检查显示既往的骨髓增生异常综合征(MDS)样特征,表现为骨髓增生活跃(粒红比:4.52:1)及显著的巨核细胞增生(图1B、C)。这些巨核细胞呈现多种异常,表现出发育异常及成熟异常征象(图1D)。骨髓活检免疫组织化学(IHC)显示CD34、CD117、髓过氧化物酶(MPO)及溶菌酶阳性表达。研究人员对骨髓来源DNA进行了髓系肿瘤靶向二代测序(NGS)panel检测。平均测序深度为 1000×,>95% 的目标区域覆盖度 ≥500×。该panel未检测到致病性或可能致病性变异。鉴于患者复杂的临床情况,进一步行全外显子测序(WES)以排查潜在遗传病因。WES分析发现新的复合杂合PIEZO1变异,具体为NM_001142864.4:c.6622A>G(p.Ile2208Val)和NM_001142864.4:c.3160C>A(p.Leu1054Met)。父母Sanger测序验证证实这些变异为复合杂合遗传,其中NM_001142864.4:c.6622A>G变异来自母亲,NM_001142864.4:c.3160C>A变异来自父亲(图1E、F)。患者父母均体健,无血液系统疾病史。经基因检测确诊后,患者被诊断为脱水性遗传性口形红细胞增多症(DHS),这解释了该患者所观察到的长期贫血、血小板增多及MDS样特征。
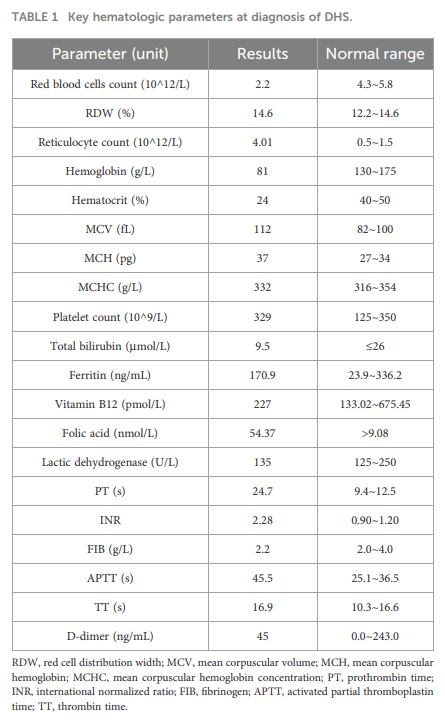
▲表1 诊断DHS时的关键血液学参数
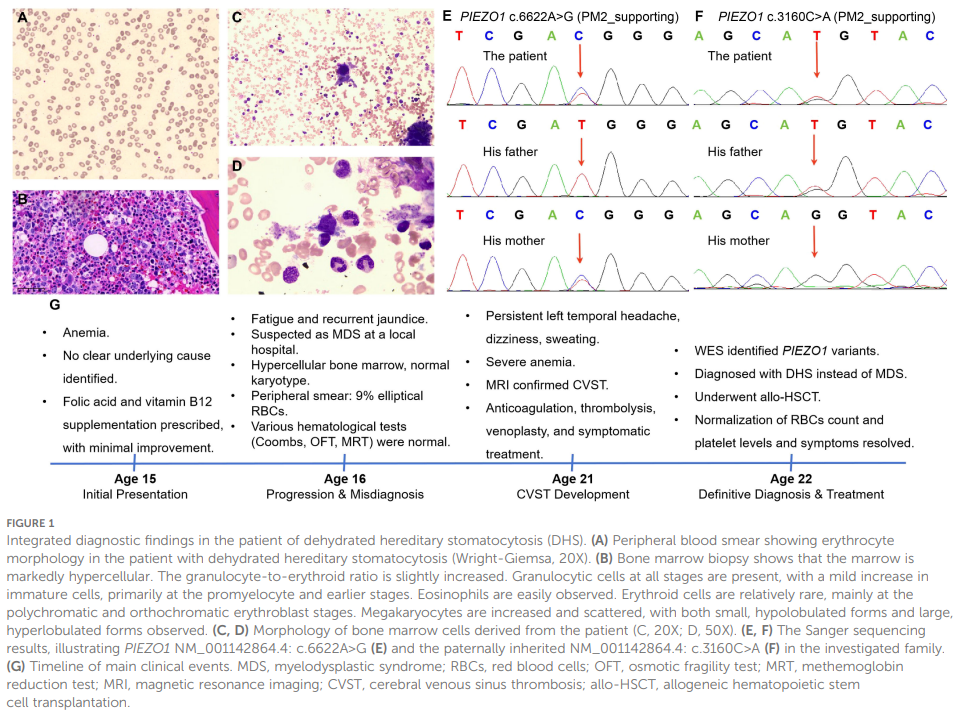
▲图1 脱水型遗传性口形红细胞增多症(DHS)患者的综合诊断结果
治疗结局和随访
患者成功接受了异基因造血干细胞移植(allo-HSCT),其血液学指标显著改善。移植后,患者的红细胞计数和血小板水平恢复正常,疲劳和黄疸症状缓解(图1G)。曾为其临床病程带来并发症的脑静脉窦血栓形成(CVST)在移植后未再复发。患者目前正在接受定期随访,以监测是否出现潜在并发症或复发。
讨 论
本病例强调了罕见血液疾病在临床特征与多种潜在诊断重叠时的诊断复杂性。该患者为22岁男性,有长期贫血病史及骨髓增生异常综合征(MDS)样特征,最终被诊断为脱水性遗传性口形红细胞增多症(DHS),其病因为复合杂合PIEZO1变异。这些变异包括 exon 45 的一个新错义变异(p.Ile2208Val)和 exon 22 的另一个错义变异(p.Leu1054Met),被确定为患者持续血液系统异常的根本原因。据研究人员所知,这两种变异均为PIEZO1基因中的首次报道。在人群数据库(gnomAD v4.0)中也未发现 c.6622A>G(p.Ile2208Val)和 c.3160C>A(p.Leu1054Met)变异。评估这些变异对蛋白质功能影响的实验证据可能不足,计算预测对其对蛋白质结构或功能的影响也存在矛盾评估。因此,根据美国医学遗传学与基因组学学会(ACMG)指南,这两种变异均被归类为意义未明变异(VUS;PM2支持证据)。重要的是,患者父母为无症状携带者,这可能提示该复合杂合对中的其中一种变异不具有致病性。虽然不能排除外显率降低或表现度变异的可能性,但复合杂合状态更可能是观察到的临床表现的原因。需要进一步的功能研究来阐明这些变异的致病性及基因型-表型相关性。
患者的持续贫血和MDS样骨髓表现(包括增生活跃及粒红比增加)提示MDS。然而,原始细胞缺失、缺乏染色体异常及荧光原位杂交(FISH)结果阴性最终排除了MDS作为确定性诊断的可能。DHS的诊断可能更具挑战性,尤其是当特征性口形红细胞罕见或在外周血涂片中难以检测时(本病例中仅观察到小部分具有口形红细胞特征的异常红细胞,导致初始诊断困难)。渗透梯度激光衍射法(一种在不同渗透条件下测量红细胞变形性的技术)是确诊DHS的有价值且通常关键的诊断工具。在DHS中,该技术通常显示渗透压曲线左移(提示红细胞脱水);但未开展此项检查,这为诊断过程带来了挑战 通过全外显子测序(WES)发现PIEZO1突变对于解决诊断歧义及确立DHS诊断至关重要——这一诊断解释了患者观察到的血液系统异常及血栓倾向。值得注意的是,基因检测不仅解决了诊断不确定性问题,还强调了在不明原因贫血及模糊骨髓表现病例中考虑遗传性红细胞疾病的重要性。鉴于对血液系统疾病种系易感性理解不断深入,近期研究强调,对疑似MDS的年轻患者,诊断检查中需纳入胚系遗传评估。扩大全面基因检测的可及性并提高对其诊断价值的认识,对于防止误诊及确保这些患者获得适当管理至关重要。
本患者脑静脉窦血栓形成(CVST)事件的发生反映了溶血性疾病中已充分证实血栓前风险。DHS以红细胞脱水和慢性溶血为特征——两者均导致高凝状态。持续溶血从血小板及其他细胞来源释放二磷酸腺苷(ADP),进而促进血小板聚集和血栓形成。此外,溶血导致一氧化氮(NO)耗竭(NO作为抑制血小板活化及促进血管舒张关键介质)。NO生物利用度降低进一步加重内皮功能障碍,为血栓形成创造有利环境。DHS中CVST的管理因血栓风险与基础溶血性疾病之间的复杂相互作用而极具挑战性。本病例中,及时启动抗凝治疗及导管导向血管内溶栓治疗取得了良好结局,且无血栓事件复发。但需密切监测潜在出血并发症,尤其是重度贫血及血小板减少患者。
异基因造血干细胞移植(allo-HSCT)在DHS中虽罕见,但对重度难治性疾病患者具有治愈潜力。本患者行HSCT的决策基于全面风险获益分析,影响决策的关键因素包括①患者年轻(22 岁)——或更能耐受HSCT相关风险(如移植物抗宿主病[GVHD]及感染);②缺乏有效替代治疗——因血栓风险,脾切除术在DHS中为禁忌证,单纯支持治疗无法解决基础遗传缺陷;③存在输血依赖型贫血——严重影响患者日常功能及生活质量。在本患者中,移植实现了血液系统异常完全缓解及血栓并发症停止,凸显了该方法的有效性。尽管HSCT存在固有风险,但在此背景下治愈的获益超过了潜在不良结局,强调了个体化评估(平衡疾病严重程度基因型及患者特异性因素)的重要性。
总之,本病例凸显了罕见血液疾病中临床特征重叠带来的诊断挑战,并强调了在病因不明的持续血液系统异常病例中全面遗传评估的重要性。DHS虽罕见,但在慢性溶血性贫血鉴别诊断中应予以考虑——尤其是患者存在血栓病史、口形红细胞或伴MDS样特征的难治性贫血时。本报告还扩展了DHS基因型谱,描述了两种新的PIEZO1变异,进一步揭示了该疾病潜在遗传多样性。
参考文献:
Sun Y, Wang T, Wang S, Chen Y, Xu Z, Shi C, Wang Z, Ouyang G. Novel compound heterozygous PIEZO1 variants in dehydrated hereditary stomatocytosis initially suspected as myelodysplastic syndromes: a case report. Front Oncol. 2025 May 21;15:1574518. doi: 10.3389/fonc.2025.1574518. PMID: 40469180; PMCID: PMC12133789.

