NGS检测助力首例携带FUS::KLF17融合的「假腺性神经鞘瘤」诊断,拓宽FUS重排相关肿瘤范围
时间:2025-09-28 12:14:24 热度:37.1℃ 作者:网络
背 景
本文报告一例 51 岁男性臂丛神经假腺性神经鞘瘤(Pseudoglandular Schwannoma,也译作假腺样神经鞘瘤)病例,该肿瘤包含神经鞘瘤常见的分子异常(22 号染色体长臂缺失,包含NF2和LZTR1基因)以及FUS::KLF17重排。假腺样神经鞘瘤是一种罕见的神经鞘瘤形态学亚型,其特征为存在腺样腔隙,内衬S100阳性、细胞角蛋白阴性的假柱状神经鞘细胞。涉及FUS和EWSR的融合常见于骨和软组织的肌上皮肿瘤。尽管涉及FUS和EWSR融合的肿瘤相对广泛,但据研究人员所知,尚未有神经鞘瘤(更不用说形态学上独特的假腺样神经鞘瘤)存在FUS重排的病例报道。因此,本病例扩展了FUS重排肿瘤的范围,强调了记录类似病例以理解这种组合的临床意义的必要性。

▲摘自《良、恶性外周神经鞘膜肿瘤新类型和少见亚型的病理诊断》
病 例
患者男,51 岁,因可触及的左侧胸部/腋窝肿块就诊。胸部MRI显示胸大肌外侧有一个 29×22×20 mm的T2加权像不均匀高信号、以边缘强化为主的肿块(图S1)。该肿块下部成分呈更明显的实性强化。肿块从胸大肌完整切除并送常规病理检查。大体检查显示,切除标本为一块 4.0 cm的不规则肌肉组织。横断面可见一个 2.4×1.3×1.2 cm边界清楚的棕黄色肿块。显微镜下,肿块边界清楚,呈实囊性、双相性,以富细胞区为主,可见密集排列的短梭形细胞束;散在分布并混杂有细胞稀疏的局灶黏液样区;未见明确的Verocay小体(图1)。病变还包含囊性腔隙和分支状腺样结构,内衬多层立方细胞,偶见腔内充满嗜酸性分泌物样物质(图1)。未见坏死、细胞学异型性或多形性,核分裂象罕见(2 个/2 mm²)。肿瘤周边可见散在淋巴滤泡。免疫组化显示,梭形细胞间质和假腺样成分均弥漫强表达S100、Sox10和p16(图2)。假腺样结构内衬细胞和间质细胞中细胞角蛋白AE1/AE3和CAM5.2亦呈局灶弱阳性(图2)。CK7、p63、EMA、SMA、calponin、结蛋白、肌细胞生成素、CD34、Glut1均不表达;增殖指数低(Ki‐67局灶最高达 5%)。
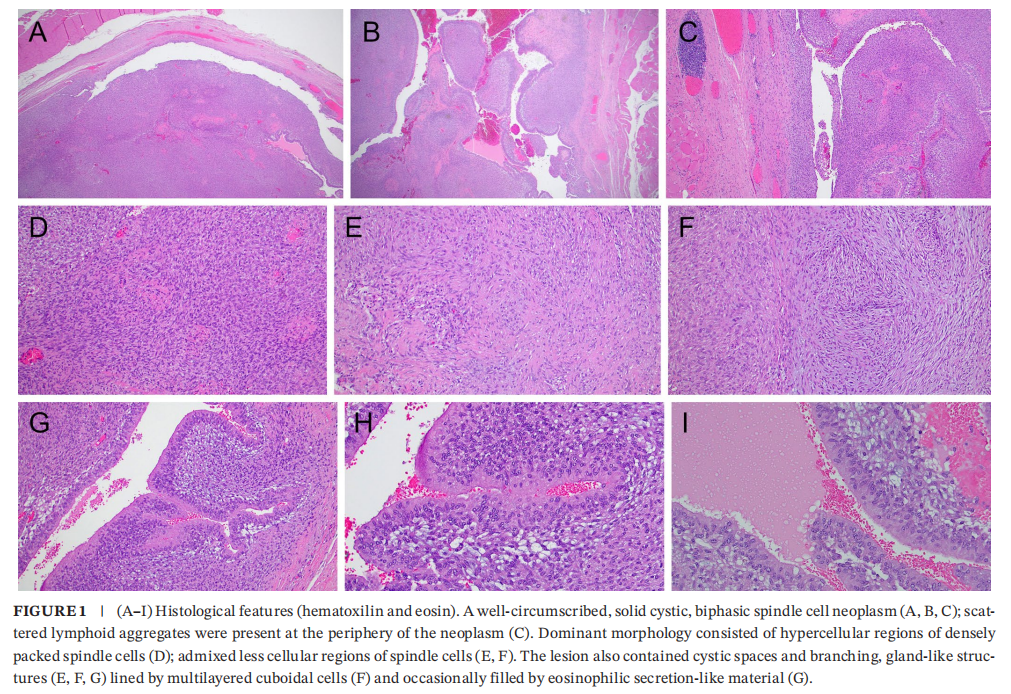
▲图1 组织学特征
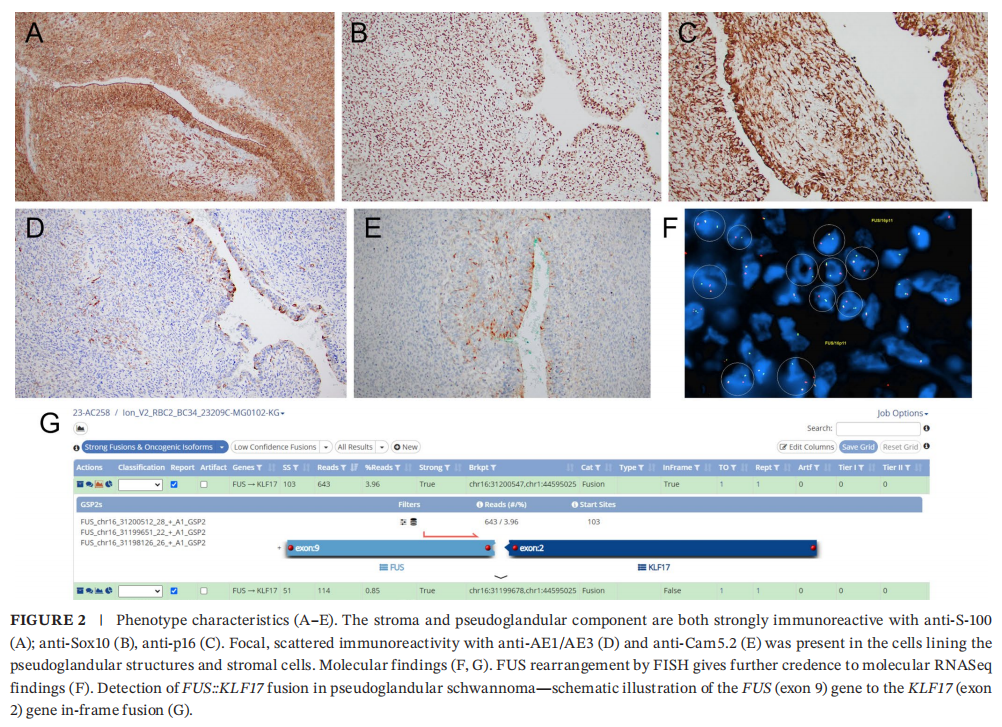
▲图2 表型特征
鉴别诊断考虑包括神经鞘瘤(周围神经鞘——神经鞘瘤或混合性神经鞘瘤)和软组织肌上皮肿瘤。送检下一代测序检测单核苷酸变异、拷贝数变异、微卫星不稳定性和肿瘤突变负荷。分子检测有几项发现,包括22号染色体q11.21–q13.2缺失(包含NF2和LZTR1基因)、IGF2基因扩增,以及由t(1;16)易位导致的FUS::KLF17融合(图2)。该肿瘤微卫星稳定,肿瘤突变负荷低。使用FUS双断裂探针(16p11)进行的荧光原位杂交(FISH)显示易位阳性(图2),为下一代测序报告的发现提供了进一步验证。使用EWSR1(22q12)双色断裂探针进行的FISH显示易位阴性。基于形态学、免疫表型和分子检测结果,诊断为假腺性神经鞘瘤,可能起源于臂丛神经。术后 12 个月,患者无复发迹象。
讨 论
神经鞘瘤是起源于施万细胞的良性、生长缓慢的肿瘤,可散发性发生,或与2型神经纤维瘤病(NF2)及神经鞘瘤病相关。它们可发生于全身各处,但常见起源部位为头颈部皮肤及皮下组织的周围神经,以及四肢屈侧表面。神经鞘瘤的发生与22号染色体q12区NF2基因的双等位基因失活有关,该基因编码肿瘤抑制蛋白——merlin。这种双等位基因失活可散发性发生,或在NF2患者中由胚系和体细胞NF2基因突变共同导致。在神经鞘瘤病(特征为多发性神经鞘瘤,通常发病较晚且不累及前庭神经)中,SMARCB1或LZTR1基因的胚系突变较为常见。这些病例的肿瘤发生过程也涉及NF2基因突变。组织学上,肿瘤经典表现为Antoni A区和Antoni B区交替的双相模式,有时在Antoni A区可见Verocay小体。《WHO肿瘤分类(第5版)》中定义了五种神经鞘瘤亚型:陈旧性、富于细胞性、丛状、上皮样及微囊性/网状型。
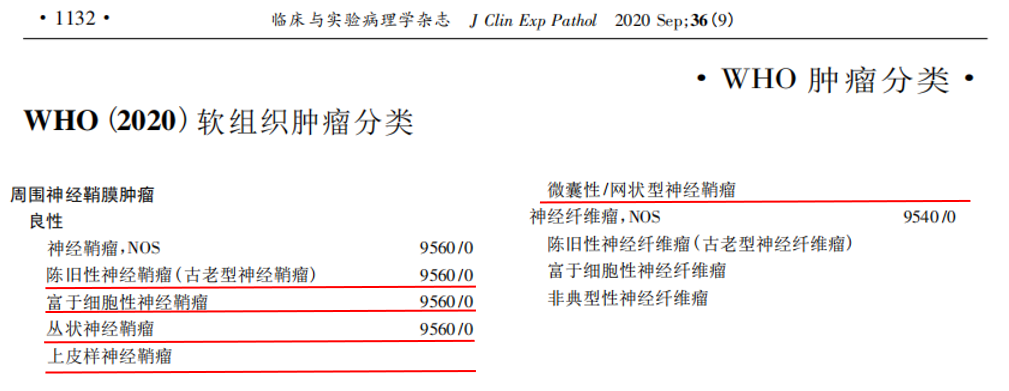
▲摘自《WHO(2020)软组织肿瘤分类》
假腺性神经鞘瘤包含腺样或囊性腔隙,内衬假柱状或立方状肿瘤性施万细胞,腔内可含有分泌物样物质,并可能存在腔内及含铁血黄素的巨噬细胞。这些假腺样腔隙的内衬细胞S100阳性,可能GFAP阳性,但始终细胞角蛋白阴性(除 1 例马尾神经病例报告中存在轻微局灶阳性外)。这与神经鞘瘤中有时可陷入的真性腺体成分不同,后者S100阴性,EMA和细胞角蛋白阳性。本例中,假腺样腔隙和间质细胞显示局灶弱阳性的细胞角蛋白AE1/AE3和Cam5.2免疫反应,而其他上皮标志物(CK7、EMA)均为阴性。
由于 2000 年之前仅报道过少数具有假腺样特征的神经鞘瘤病例,因此该亚型被认为罕见。然而,Robins对 202 例神经鞘瘤的回顾性研究发现,16 例(7.9%)神经鞘瘤具有假腺样特征。Ud Din等人对 971 例神经鞘瘤进行了类似回顾,报道 61 例(6.3%)存在假腺样成分,表明该特征可能比之前认为的更常见。两位作者均认为假腺样腔隙是Verocay小体囊性变的结果,但尚未有对具有假腺样成分的神经鞘瘤进行分子特征分析的尝试。本例中未观察到典型的、结构完整的Verocay小体。
另一个鉴别诊断考虑为混合性神经鞘瘤(神经鞘瘤合并神经束膜瘤或神经鞘瘤合并神经纤维瘤),但EMA、Glut1和CD34免疫反应阴性,排除了神经束膜瘤和神经纤维瘤成分。
常见存在EWSR或FUS融合(包括本例的FUS::KLF17融合)的肿瘤类型是肌上皮肿瘤(METs),这是一组临床和形态学上具有多样性的肿瘤,可发生于涎腺、皮肤或软组织。肌上皮肿瘤可包含导管结构,良恶性不等,不同部位定义恶性的命名和特征有所差异。目前尚无肌上皮分化的特异性免疫染色,但此类肿瘤通常共表达上皮标志物(如细胞角蛋白和EMA)以及S100。它们还可能表达钙调蛋白(calponin)、GFAP、SMA、p63、SOX10。本例中,其他相关肌上皮免疫标志物(SMA、calponin、p63)均为阴性;EWSR荧光原位杂交(FISH)也为阴性,因此肌上皮肿瘤的诊断可能性较低。
尽管骨/软组织/内脏与皮肤/涎腺的肌上皮肿瘤在形态学和免疫表型上存在重叠,但其遗传学特征差异显著。皮肤和涎腺肌上皮肿瘤常存在PLAG1和HMGA2基因重排。骨和软组织肌上皮肿瘤最常见的是存在EWSR融合,较少见的是FUS基因融合。已报道的EWSR融合伙伴众多,包括POU5F1、PBX1、PBX3、ZNF444、KLF15、KLF17和ATF1。FUS最常见的融合伴侣是KLF17;也有FUS::POU5F1融合的病例报道。
FUS和EWSR属于TET家族,编码核RNA/DNA结合蛋白,主要参与RNA代谢、DNA修复以及转录、剪接、mRNA转运等细胞过程。这些基因在肌上皮肿瘤和其他多种肿瘤中可作为易位伙伴相互替代,包括黏液样脂肪肉瘤(FUS/EWSR::DDIT3)、低级别纤维黏液样肉瘤(FUS/EWSR::CREB3L1或CREB3L2)、血管瘤样纤维组织细胞瘤(FUS/EWSR-ATF1)、尤因肉瘤,以及其他肉瘤和部分血液系统恶性肿瘤。肌上皮肿瘤中FUS最常见的融合伙伴是KLF17,后者是一种转录调节因子,可激活TGF-β/SMAD信号通路,该通路通常抑制肿瘤生长和转移。易位断点位于KLF17的5'非翻译区,因此推测其可能破坏KLF17的翻译或功能。
据研究人员所知,FUS/EWSR重排肿瘤谱此前不包括神经鞘瘤。本病例的特殊之处在于,它是一例具有假腺样特征的细胞性神经鞘瘤(一种罕见的形态变异型),同时包含FUS::KLF17融合以及神经鞘瘤中常见的22q缺失。这种组织学特征与分子学发现的组合此前尚未见报道,扩展了FUS相关肿瘤范围,并强调了分子检测在罕见肿瘤诊断和理解中的重要性。
结 论
本文报告一例极为罕见的具有假腺样特征的细胞性神经鞘瘤病例,该肿瘤存在预期的22号染色体长臂缺失(包含NF2和LZTR1基因)以及FUS‐KLF17融合。假腺样成分的存在(其特征为S100阳性、由神经鞘细胞内衬的腺样结构)代表了一种罕见的神经鞘瘤形态学变异型。FUS‐KLF17融合的共存值得关注,因为这种遗传异常主要报道于肌上皮肿瘤,而在神经鞘瘤中从未见报道。据研究人员所知,这种组合此前尚未见报道;因此,本病例扩展了FUS相关肿瘤范围。进一步研究和记录类似病例对于理解这种组合的临床意义至关重要。
实体瘤1560基因融合RNA检测(NGS方法学)项目,可检测FUS::KLF17融合在内超过1560种已知融合和数百个基因相关的未知融合。实体瘤染色体拷贝数变异检测(CNV-Seq方法学)项目,可检测22q缺失,辅助临床诊疗。
参考文献:
Givi J, Wu D, Bakkar R, Afkhami M, Bell D. Pseudoglandular Schwannoma With FUS::KLF17 Fusion: Broadening the Spectrum of FUS-Associated Tumors. Genes Chromosomes Cancer. 2025 Aug;64(8):e70077. doi: 10.1002/gcc.70077. PMID: 40878925; PMCID: PMC12340572.

